Ky shkrim flet për një grup gjendjesh gjenetike të quajtura talasemi dhe përfshin detaje të simptomave, shkaqeve dhe trajtimeve dhe testeve të disponueshme.
Përmbledhje
- Talasemia është një gjendje e zakonshme e trashëguar e qelizave të kuqe të gjakut që mund të shkaktojë anemi gjatë gjithë jetës, shpesh kërkon transfuzione të rregullta të gjakut dhe/ose medikamente.
- Ekzistojnë dy lloje kryesore: talasemia alfa (α) dhe talasemia beta (β).
Shkaktohet nga ndryshimet në gjenet alfa globin ose beta globin që kodojnë për hemoglobinën e proteinave që mbart oksigjen në qelizat e kuqe të gjakut. - Talasemia trashëgohet në mënyrë autosomale recesive dhe mbartësit gjenetikë të talasemisë zakonisht nuk kanë shenja apo simptoma.
- Çiftet ku të dy partnerët janë mbartës gjenetikë të të njëjtit lloj talasemie kanë probabilitet 1 në 4 (25%) për të pasur një fëmijë me talasemi.
Çfarë është talasemia?
Talasemia është një gjendje e zakonshme e trashëguar e qelizave të kuqe të gjakut që shkakton anemi gjatë gjithë jetës. Njerëzit me talasemi kanë qeliza të kuqe të gjakut të vogla dhe të zbehta për shkak të nivelit të ulët të hemoglobinës (proteinë brenda qelizave të kuqe të gjakut) funksionale. Kjo njihet si anemi dhe mund të shkaktojë lodhje, lëkurë të zbehtë (zbehje) dhe komplikime të tjera serioze.
Dy llojet kryesore të talasemisë janë:
- Talasemia alfa (α): më e zakonshme tek njerëzit prejardhja e të cilëve është nga Kina, Azia Juglindore, Mesdheu Lindor, Afrika, Ishujt Paqësorë dhe Zelanda e Re (Maori).
- Talasemia beta (β): më e zakonshme tek njerëzit prejardhja e të cilëve është nga Lindja e Mesme, Mesdheu, Afrika, Nënkontinenti Indian, Azia Qendrore dhe Juglindore dhe Karaibet.
Alfa (α) Talasemia
Ekzistojnë dy forma të talasemisë alfa që mund të shkaktojnë probleme shëndetësore:
- Sëmundja e Hemoglobinës H (HbH): është një formë e butë e talasemisë alfa që shkakton anemi të butë deri të moderuar. Trajtimi mund të përfshijë transfuzione gjaku (ndonjëherë ose rregullisht). Karakteristika të tjera përfshijnë shpretkë (organi që filtron gjakun) të zmadhuar dhe zverdhje të syve dhe lëkurës.
- Sindroma Hemoglobin Barts hidrop fetal (Hb Barts): është një formë e rëndë e alfa talasemisë ku lëngjet e tepërta grumbullohen tek foshnja në zhvillim gjatë shtatzënisë për shkak të anemisë së rëndë. Foshnja zakonisht nuk mbijeton shumë kohë pas lindjes.
Mund të ketë gjithashtu komplikime për nënën shtatzënë që mban një fëmijë të prekur, përfshirë presionin e lartë të gjakut (preeklampsia). Është gjithashtu e mundur që të jeni një mbartës gjenetik për alfa talaseminë, i njohur si tipari alfa plus i talasemisë ose tipari alfa zero i talasemisë, por asnjëra nga këto e vetme nuk ka të ngjarë të paraqesë ndonjë problem të rëndësishëm shëndetësor.
Beta talasemia
Ekzistojnë dy forma të beta talasemisë që mund të shkaktojnë probleme shëndetësore:
- Beta Talasemia e ndërmjetme: është një version më i butë i beta talasemisë major, duke shkaktuar anemi të butë deri të moderuar. Simptomat mund të fillojnë në fëmijërinë e hershme ose më vonë gjatë jetës dhe mund të nevojiten transfuzione gjaku. Simptoma të tjera përfshijnë rritjen e ngadaltë dhe ndryshime të kockave.
- Beta Talasemia Major: e njohur edhe si anemia Cooley është forma më e rëndë e beta talasemisë. Fëmijët zhvillojnë anemi serioze brenda vitit të parë ose të dy-të jetës dhe kërkojnë transfuzione të rregullta të gjakut gjatë gjithë jetës së tyre.
Grumbullimi i hekurit për shkak të transfuzioneve të rregullta mund të shkaktojë komplikime shëndetësore dhe nevojiten medikamente për të larguar hekurin e tepërt. Simptoma të tjera mund të përfshijnë mungesën e zhvillimit, verdhëzën (zverdhjen) e syve dhe lëkurës, zmadhimin e shpretkës, ndryshimet e kockave dhe vonesën në zhvillim.
Është gjithashtu e mundur që të jetë një mbartës gjenetik për beta talaseminë, i njohur si tipar beta talasemia ose beta talasemia minore; asnjëra nga këto, e vetme, në përgjithësi nuk paraqet ndonjë problem të rëndësishëm shëndetësor.
Çfarë shkakon talasemia?
Brenda qelizave të kuqe të gjakut gjendet proteina e hemoglobinës, puna e të cilës është të transportojnë oksigjen nga mushkëritë në të gjitha pjesët e trupit. Hemoglobina është ajo që u jep eritrociteve (qelizat e kuqe të gjakut) ngjyrën e kuqe dhe përbëhet nga katër zinxhirë, dy zinxhirë alfa globin dhe dy zinxhirë beta globin. Ndryshimet në gjenet që kodojnë për zinxhirët alfa globin dhe zinxhirët beta globin mund të bëjnë që hemoglobina të mos funksionojë siç duhet. Simptomat do të ndryshojnë në varësi të faktit nëse është zinxhiri alfa globin ose zinxhiri beta globin që nuk funksionon.
Alfa Talasemia
Ekzistojnë dy çifte gjenesh alfa globinë në kromozomin 16 (gjithsej katër gjene alfa globin, dy në secilin kromozom), që kodojnë për zinxhirët alfa globin. Talasemia alfa ndodh kur një ose më shumë nga katër kopjet e gjenit alfa globin nuk funksionojnë.
- Mbartës gjenetikë për alfa talaseminë: kanë një ose dy kopje jo funksionale të gjeneve të alfa globinës. Meqenëse ka të paktën dy kopje të gjeneve që funksionojnë, mjafton sasia e prodhuar e zinxhirëve alfa globinë për të parandaluar çdo ndërlikim serioz shëndetësor. Njerëzit me një kopje të gjenit që nuk punon kanë tipin alfa plus të talasemisë dhe mund të mos kenë ndonjë indikacion për të qenë një mbartës gjenetik të talasemisë nga testet rutinë të gjakut. Njerëzit me dy kopje të gjeneve që nuk punojnë kanë tipin alfa zero të talasemisë dhe zakonisht kanë qeliza të kuqe të gjakut të vogla dhe të zbehta në testet rutinë të gjakut.
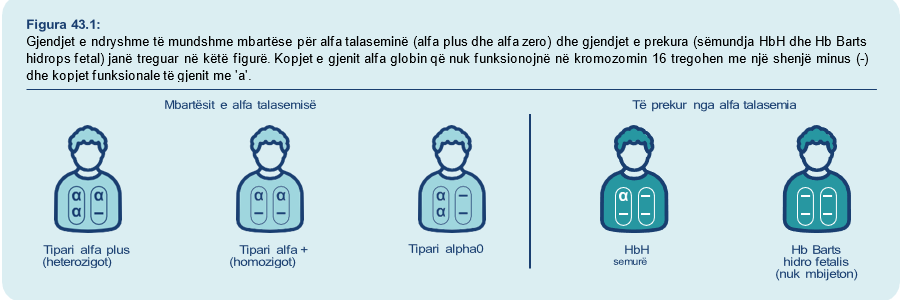
- Njerëzit me talasemi alfa: kanë tre ose katër kopje jo-funksionale të gjeneve të alfa globinës. Sëmundja e Hemoglobinës H shkaktohet nga tre kopje të gjeneve që nuk punojnë dhe shkakton një anemi të butë deri të moderuar që është e shërueshme. Hemoglobin Barts hidrops fetal është gjendja ku të katër gjenet alfa globin nuk po punojnë siç duhet dhe foshnjat me këtë zakonisht nuk mbijetojnë shumë kohë pas lindjes. Talasemitë alfa të ndryshme dhe ndryshimet e gjeneve që rezultojnë në secilën formë të talasemisë alfa janë treguar në Figurën 43.1.
Trupi ynë përbëhet nga miliarda qeliza, dhe në secilën prej tyre ka udhëzime,të quajtura gjene, që bëjnë që të gjithë përbërësit e nevojshëm strukturorë dhe kimikë të trupit të funksionojnë siç duhet. Këto gjene janë të paketuara në fije të gjata të njohura si kromozome.
Të gjithë kemi 46 kromozome të grupuara në 23 çifte. Një kopje të secilit çift e kemi trashëguar nga nëna dhe tjetrin nga babai. 22 çiftet e para të kromozomeve njihen si kromozome autosomale. Çifti i 23 -të përbëhet nga kromozomet seksuale të quajtur X dhe Y. Meshkujt kanë një kromozom X dhe një Y kurse femrat kanë dy kopje të kromozomit X.
Meqënëse të gjithë kromozomet tanë vijnë në çifte, të gjitha gjenet vijnë gjithashtu në çifte. Gjenet japin udhëzimet për proteinat, të cilat janë blloqet ndërtuese të qelizave që përbëjnë trupin tonë.
Edhe pse ne të gjithë kemi ndryshime në gjenet tona, ndonjëherë kjo mund të ndikojë në mënyrën se si trupat tanë rriten dhe zhvillohen. Në përgjithësi, variacionet e gjeneve që nuk kanë ndikim në shëndetin tonë quhen variante beninje ose polimorfizma. Këto variante kanë tendencë të jenë më të zakonshme tek njerëzit.
Më rrallë, variacionet mund të ndryshojnë gjenin duke bërë që ai të dërgojë një mesazh tjetër. Këto ndryshime mund të nënkuptojnë që gjeni nuk punon siç duhet ose punon në një mënyrë tjetër të dëmshme. Një ndryshim në një gjen që shkakton një gjendje shëndetësore ose zhvillimi quhet një variant ose mutacion patogjen.
Variantet e gjeneve mund të trashëgohen nga një prind, ose të ndodhin për herë të parë në një person, njëherë që keni një variant të gjenit, ai mund të kalojë tek brezat e ardhshëm. Kjo quhet trashëgimi gjenetike.
Beta talasemia
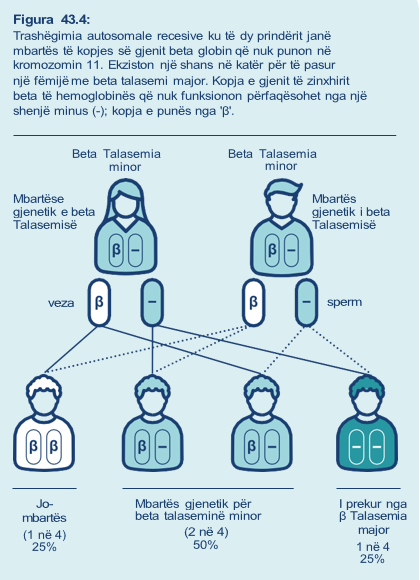
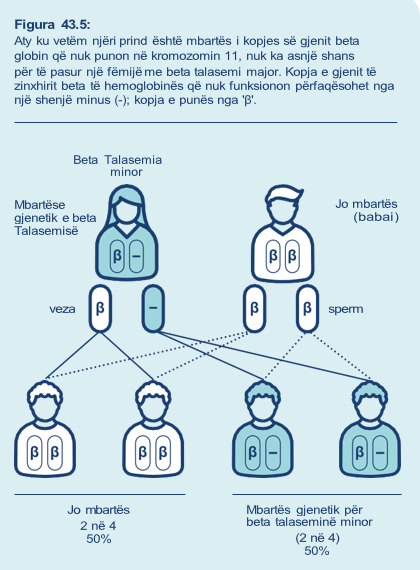
Ekzistojnë një palë gjene beta globin në kromozomin 11 (gjithsej dy gjene beta globin, një në secilin kromozom), që kodojnë për zinxhirët beta globin. Talasemia beta ndodh kur një ose të dy kopjet e kopjeve të gjenit beta globin nuk punojnë.
• Mbartësit për beta talaseminë: kanë një kopje të gjenit që nuk punon dhe një kopje funksionale të gjenit beta globin. Kjo quhet beta talasemia minor.
Mbartësit gjenetikë janë përgjithësisht të shëndetshëm, por zakonisht kanë qeliza të kuqe të gjakut më të zvogëluara dhe të zbehta.
• Personat e prekur nga beta talasemia:
Të dy kopjet e gjenit beta globin nuk ju funksionojnë. Ashpërsia e simptomave ndryshon në varësi të variacionit gjenetik dhe mund të klasifikohet si beta talasemi major ose beta talasemia e ndërmjetme.
Si trashëgohet talasemia?
Talasemia është një gjendje gjenetike që ndjek një model të trashëgimisë autosomale recesive. Gjenet e alfa dhe beta globinës janë të vendosura në kromozomet autosomalë (16 dhe 11), dhe për këtë arsye prek meshkujt dhe femrat në mënyrë të barabartë. Recesive do të thotë që, për të zhvilluar shenja dhe simptoma të gjendjes, të dyja kopjet e gjeneve e globinës duhet të mos punojnë (ose të paktën 3 nga 4 gjenet alfa globinës).
Për Alfa Talaseminë:
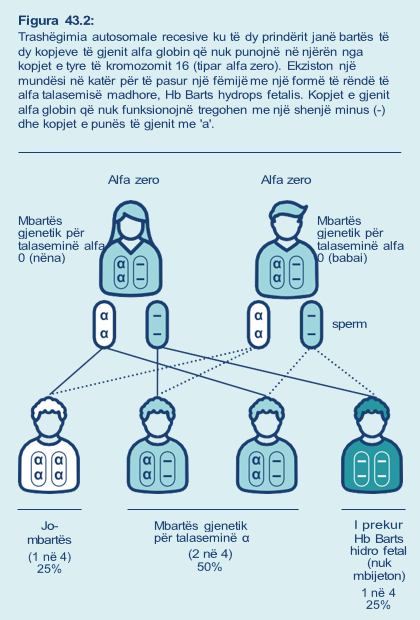
Nëse në një çift janë të dy mbartës gjenetikë të tiparit alfa zero të talasemisë (Figura 43.2), në çdo shtatzëni ka:
- 1 në 4 (25%) mundësi që ata të kenë një fëmijë i cili trashëgon të dy kopjet e variantit të gjenit recesiv nga prindërit e tyre. Në këtë rast, nuk do të prodhohet asnjë produkt gjenesh funksionalë dhe fëmija i tyre do të ketë Hb Barts hidrops fetal dhe nuk do të mbijetojë.
- 1 në 4 (25%) shans që fëmija i tyre të trashëgojë të dy kopjet e gjenit funksional dhe të mos manifestojë alfa talasemi apo të jetë mbartës gjenetik.
- 1 në 2 (50%) mundësi që fëmija i tyre të trashëgojë variantin e gjenit recesiv dhe kopjen e gjenit nga prindërit por të jetë mbartës gjenetik, pra i paprekur për talaseminë alfa, ashtu si prindërit.
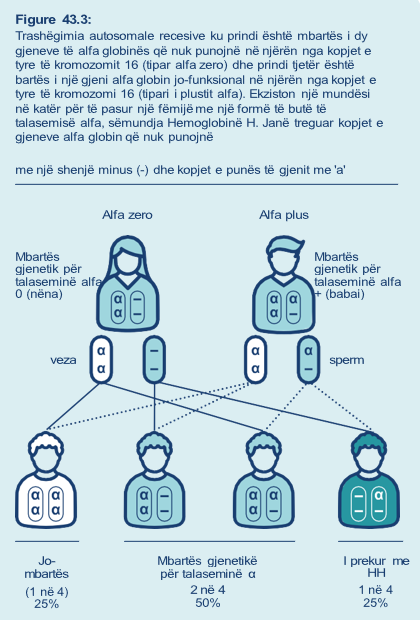
Nëse një çift ku njëri prind është mbartës i tipit alfa zero të talasemisë dhe prindi tjetër është mbartës i tiparit alfa plus talasemia (Figura 43.3), në çdo shtatzëni ka:
- 1 në 4 (25%) mundësi që ata të kenë një fëmijë i cili trashëgon të dy kopjet e variantit të gjenit recesiv nga prindërit e tyre. Në këtë rast, do të krijohet proteina funksionale në sasi të reduktuar dhe fëmija i tyre do të ketë sëmundjen Hemoglobinë H
- 1 në 4 (25%) shanset që fëmija i tyre të trashëgojë të dy kopjet e gjenit funksional dhe të mos ketë alfa talasemi dhe të mos jetë mbartës gjenetik
- 1 në 2 (50%) mundësitë që fëmija i tyre të trashëgojë variantin e gjenit recesiv dhe kopjen e gjenit nga prindërit dhe ata të jenë mbartës gjenetik i paprekur për alfa talaseminë, ashtu si secili prind.
Nëse në një çift janë të dy mbartës gjenetikë të alfa plus tiparit të talasemisë, atëherë, në rastin më të keq, fëmija ka një 1 në 4 shans (25%) për të trashëguar të dy variantet e vetme të gjenit, një në secilin kromozom.
Meqenëse ka ende dy kopje të gjenit alfa globin, proteina e mjaftueshme e hemoglobinës është bërë për të parandaluar ndonjë problem të rëndësishëm shëndetësor. Prandaj foshnja do të jetë një mbartës i shëndetshëm gjenetik.
Po kështu, nëse vetëm njëri prind është mbartës i tiparit alfa zero të talasemisë ose tipar alfa plus talasemia, atëherë në rastin më të keq foshnja ka shans 1 në 2 (50%) të jetë gjithashtu një mbartës gjenetik. Në të gjitha këto raste, foshnja nuk do të ketë talasemi alfa.
Nëse një çift ku njëri prind është mbartës i tipit alfa zero të talasemisë dhe prindi tjetër është mbartës i tiparit alfa plus talasemia (Figura 43.3), në çdo shtatzëni ka:
- 1 në 4 (25%) mundësi që ata të kenë një fëmijë i cili trashëgon të dy kopjet e variantit të gjenit recesiv nga prindërit e tyre. Në këtë rast, do të krijohet proteina funksionale në sasi të reduktuar dhe fëmija i tyre do të ketë sëmundjen Hemoglobinë H
- 1 në 4 (25%) shanset që fëmija i tyre të trashëgojë të dy kopjet e gjenit funksional dhe të mos ketë alfa talasemi dhe të mos jetë mbartës gjenetik
- 1 në 2 (50%) mundësitë që fëmija i tyre të trashëgojë variantin e gjenit recesiv dhe kopjen e gjenit nga prindërit dhe ata të jenë mbartës gjenetik i paprekur për alfa talaseminë, ashtu si secili prind.
Nëse në një çift janë të dy mbartës gjenetikë të alfa plus tiparit të talasemisë, atëherë, në rastin më të keq, fëmija ka një 1 në 4 shans (25%) për të trashëguar të dy variantet e vetme të gjenit, një në secilin kromozom. Meqenëse ka ende dy kopje të gjenit alfa globin, proteina e mjaftueshme e hemoglobinës është bërë për të parandaluar ndonjë problem të rëndësishëm shëndetësor.
Prandaj foshnja do të jetë një mbartës i shëndetshëm gjenetik. Po kështu, nëse vetëm njëri prind është mbartës i tiparit alfa zero të talasemisë ose tipar alfa plus talasemia, atëherë në rastin më të keq foshnja ka shans 1 në 2 (50%) të jetë gjithashtu një mbartës gjenetik. Në të gjitha këto raste, foshnja nuk do të ketë talasemi alfa.
Çfarë ndodh nëse një mbartës alfa talasemie dhe një mbartës talasemie beta kanë një familje?
Është e mundur që njëri prind të jetë mbartës gjenetik i alfa talasemisë dhe prindi tjetër të jetë mbartës gjenetik i beta talasemisë. Ekziston një shans në 4 (25%) që fëmija të trashëgojë të dy variantet gjenetike dhe të jetë mbartës i talasemisë alfa dhe beta talasemisë. Meqenëse gjenet e përfshira janë të ndryshme dhe kanë role të ndryshme në prodhimin e hemoglobinës, foshnja nuk do të zhvillojë asnjë shenjë apo simptomë të anemisë, dhe do të jetë vetëm një mbartës i shëndetshëm gjenetik për të dyja kushtet.
Po variantet e hemoglobinës dhe talasemisë?
Variantet e hemoglobinës janë një term i përdorur për të përshkruar një ndryshim specifik në gjenet e globinës alfa ose beta, që ndryshojnë strukturën e hemoglobinës prodhuar, më tepër sesa reduktojnë sasinë e hemoglobinës. Disa shembuj të zakonshëm përfshijnë Hemoglobinën S (e njohur gjithashtu si sëmundja e qelizave drapër), ose Hemoglobinën E. Këto janë të dy variantet e zinxhirit beta globin.
Ndonjëherë njëri prind mund të jetë mbartës gjenetik i talasemisë dhe prindi tjetër është mbartës gjenetik i variantit të hemoglobinës. Në varësi të kombinimit, është e mundur që një fëmijë të ketë disa simptoma të ngjashme me talaseminë. Është mirë që të flitet për rezultatet e mundshme me mjekun e kujdesit shëndetësor bazuar në statusin gjenetik të vetë individit dhe të partnerit të tij.
A ka testime të aksesueshme për talaseminë?
Njerëzit me talasemi kanë anemi kronike dhe kjo mund të zbulohet përmes testeve të specializuara të gjakut. Këto teste mund të identifikojnë gjithashtu mbartës gjenetikë të talasemisë, edhe pse disa forma më të buta si alfa plus nuk gjenden vetëm nëpërmjet këtyre testeve. Testimi gjenetik i gjeneve alfa globin ose gjeni beta globin mund të konfirmojë një diagnozë të talasemisë ose statusit të mbartësit gjenetik të një personi.
Testimi për statusin e mbartësit gjenetik
Kur një person zbulohet se është një mbartës gjenetik i talasemisë, të afërmit e tyre të shkallës së parë (prindërit, fëmijët, vëllezërit dhe motrat) kanë të gjithë një shans 1 në 2 (50%) për të qenë gjithashtu mbartës gjenetik. Ekzaminimi me anë të testeve të specializuara të gjakut përmes mjekut tuaj të familjes ose testimi gjenetik mund të ofrohet nëse variantet e gjeneve janë identifikuar në familjen tuaj.
Planifikimi i një shtatzënie
Për çiftet që janë të dy mbartës gjenetikë të talasemisë, testimi duhet të jetë i disponueshëm gjatë shtatzënisë, duke dhënë informacion nëse fëmija nuk është i prekur, ka talasemi ose është një mbartës gjenetik i talasemisë. Testimi gjenetik në një shtatzëni për talaseminë është opsional dhe duhet të flitet hollësisht me mjekun, maminë ose këshilltarin gjenetik.
Ka mundësi gjithashtu të realizohet dhe diagnoza gjenetike para implantimit (PGD) për të ditur nëse një embrion i krijuar nëpërmjet fekondimit in vitro (FIV) është i prekur nga talasemia. Kur planifikonhet një familje, opsionet për testimin gjenetik më së miri duhet të fliten dhe të konsiderohen përpara shtatzënisë.